Leire Torices, Javier de las Heras, Juan Carlos Arango-Lasprilla, Jesús M. Cortes, Caroline E. Nunes-Xavier , Rafael Pulido. MMADHC premature termination codons in the pathogenesis of cobalamin D disorder: Potential of translational readthrough reconstitution. Molecular Genetics and Metabolism Reports. In press, 2021 [pdf]
Abstract
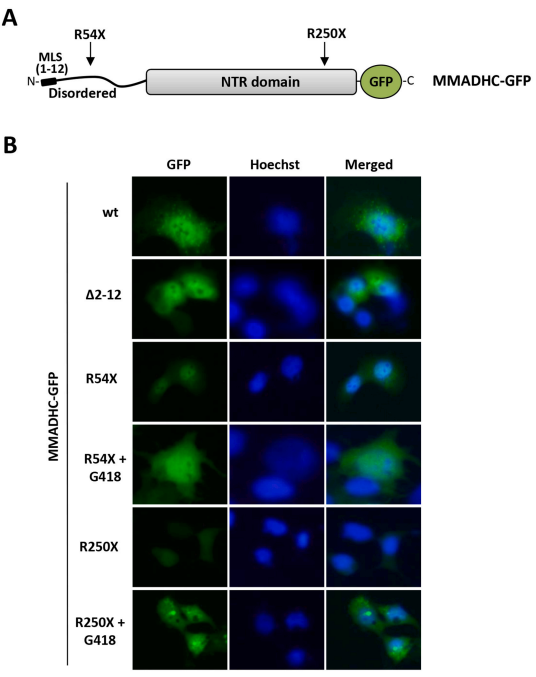
Mutations in the MMADHC gene cause cobalamin D disorder (cblD), an autosomal recessive inborn disease with defects in intracellular cobalamin (cbl, vitamin B12) metabolism. CblD patients present methylmalonic aciduria (MMA), homocystinuria (HC), or combined MMA/HC, and usually suffer developmental delay and cognitive deficits. The most frequent MMADHC genetic alterations associated with disease generate MMADHC truncated proteins, in many cases due to mutations that create premature termination codons (PTC). In this study, we have performed a comprehensive and global characterization of MMADHC protein variants generated by all annotated MMADHC PTC mutations in cblD patients, and analyzed the potential of inducible translational PTC readthrough to reconstitute MMADHC biosynthesis. MMADHC protein truncation caused by disease-associated PTC differentially affected the alternative usage of translation initiation sites, protein abundance, and subcellular localization of MMADHC. Aminoglycoside compounds induced translational PTC readthrough of MMADHC truncated variants, allowing the biosynthesis of full-length MMADHC in a PTC-specific manner. Our results suggest that translational PTC readthrough-based interventions could complement current therapies for cblD patients carrying specific MMADHC PTC mutations.